
What is Bioinformatics?
Bioinformatics is an emerging discipline which combines the knowledge of information science, mathematics and biology. Bioinformatics plays a crucial role in answering important biological questions by aiding in analysis of genome and gene expression. With the help of bioinformatics, we are able to annotate the genome which has allow us to understand the function and also, we are able to know the root cause of many diseases, identify specific cell types and developed medication as per the patients’ profile. In addition to this, bioinformatics has wide array of tool for analysis of protein structure and function, making us able to understand important biological pathways and interaction of different molecules.
Due to use of large amount of computer resources, softwares and algorithms in life sciences research, it is also described as computational biology. Bioinformatics has three main components which include:
Creation of database
Development of softwares and tool
Analysis of biological data by employing a suitable tool or software
Database
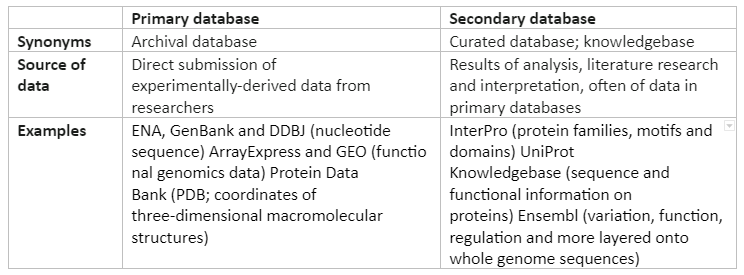
As the biological methods are being improved, a great amount of biological data is being produced. In order to utilize this data in a better way, efficient sorting and organization of this data is a necessity, this is done with the help of multiple websites which accepts data from various sources and then by using different algorithms organize them into a database. There are hundreds of different bioinformatics algorithms that have been established either with general features or with specific features. The databases can be categorized into two types:
Primary databases: the databases that have original sequence data e.g., GenBank (NCBI), the Nucleotide Sequence Database (EMBL), and the DNA Databank of Japan (DDBJ). There is an exchange of data between these databases on a daily basis.
Secondary database: These databases have derived data taken from the primary database. For instance, PROSITE, PRINTS, and InterPro are protein databases.
Besides these, there are also some biological databases which consist of catalogs of human diseases like Online Mendelian Inheritance in Man (OMIM). Some of the aspects of primary and secondary databases is given in Table 1:
Table 1: Aspect of Primary and Secondary Databases
Tools and Analysis
There are many bioinformatics tools that have been developed for analysis. These tools are based on different algorithms to give an optimized analysis as per the need of the researcher. Without the bioinformatics tools, it is not easy to handle large amounts of data and organize it for analysis manually, as it is prone to many human errors.
The main objective of the tools is analysis. For instance, if we want to do a pairwise sequence alignment, we can use Emboss NEEDLE. NEEDLE simply allows the global sequence alignment of two input sequences. For the same purpose Emboss WATER can be used.
Similarly, many tools were developed for multiple sequence alignment like “Clustal W” and “Clustal Omega”. These tools are able to align multiple input sequences and determine conserved bases which is quite useful for phylogenetic analysis. Besides these, a large number of tools are available for evolutionary biology and phylogenetic studies like UPGMA, and Neighbor joining methods etc. These tools are designed with specific types of algorithms and specific programming languages.
Furthermore, many tools have been specifically designed for protein analysis in which they are able to predict 3D structure of proteins. The tools develop for this purpose are I-TASSER, trRosetta, RaptorX-SS8, Jpred, etc. In addition to this the structure information of protein can be mined from protein database which has the amino acid sequence, structure and function information from protein. Important databases are PDB, SCOP and HOMSTRAD.
One of the most important analysis tools developed by bioinformatics is molecular docking tools, these tools are able to determine the interaction between two molecule which results in formation of stable adduct. This principle plays a vital role in drug discovery pathway.
Bioinformatics in Life Sciences
Life sciences includes all the branches of science that are involved in scientific study of life. Due to a large amount of research, a high amount of variety of data which includes RNA, DNA, amino acid sequences data, microarray gene expression data and protein structure, biological pathways and signals data. In the past decade, bioinformatics has emerged as the problem solver for the life science researchers by providing them with tools that can not help them to efficiently organize data but also analyze it. These tools have high coverage i.e., from genome annotation and prediction of function to gene expression analysis. The illustration of bioinformatics tools and its application is given in Figure 1:
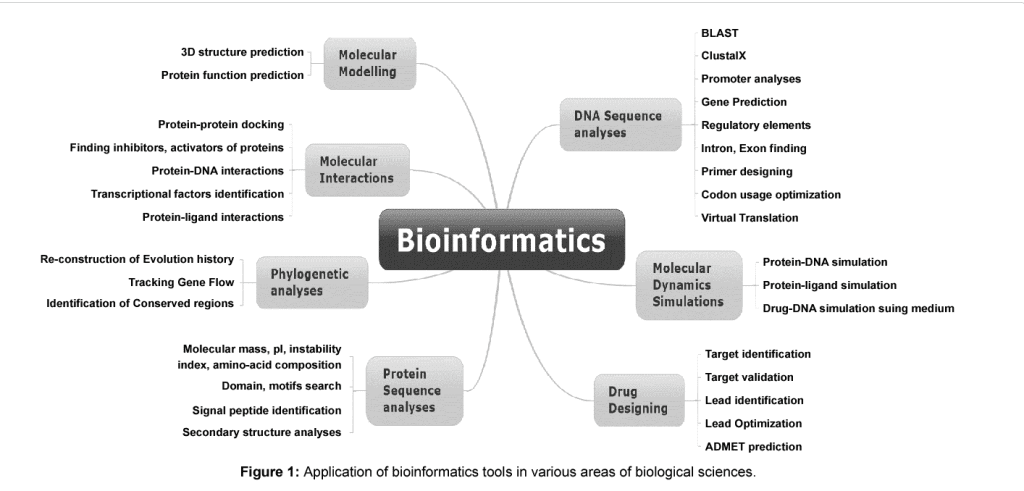
Application of Bioinformatics tool in life sciences
The details of some of the applications are given in the next section.
DNA Sequencing and analysis
Bioinformatics allows us to efficiently and rapidly sequence DNA and then it also helps in understanding features, functions and structure of the genome. The next-generation sequencing is the culmination of bioinformatics techniques. Next generation sequencing is a high-throughput sequencing technique which has ability to identify genetic variants and variable gene expression patterns that are linked with disease state. These finding are very beneficial clinically and allows to formulate a strategy against diseases. The workflow of NGS consists of four steps:
Sample Extraction: NGS can be done on any sample from which DNA or RNA can be extracted. The quality of DNA or RNA must be ensured as it is critical for further analysis.
Library preparation: In this step, with the help of PCR amplification, libraries are designed by using primer of interest which amplify specific sequences. Also, adaptor ligation step is also added to amplified fragment of DNA or RNA. These adaptors are specific oligonucleotide sequences which interacts with the sequencing flow cell surface. In cases where multiple samples are sequenced in a single step, a unique identifier is added which is afterwards ‘demultiplexed’ during analysis.
DNA sequencing: In this step, parallel sequencing is done with the help of an NGS platform. = The platforms used for NGS are Illumina (Solexa) sequencing, Roche 454 sequencing, Ion torrent Proton / PGM sequencing and SOLiD sequencing. Illumine is the most popular platform which has generated almost 90% of sequencing data.
Alignment and Data Analysis: After sequencing, specialized software is used to analyze the data. But before any analysis, the quality control step is done to ensure the quality of sequence reads. Then alignment is done with a reference genome. Finally, reads are then compared with the reference genome to identify variants link to disease etc. the analysis can also help in variant annotation by comparing with known genes and regulatory sequences.
Gene prediction
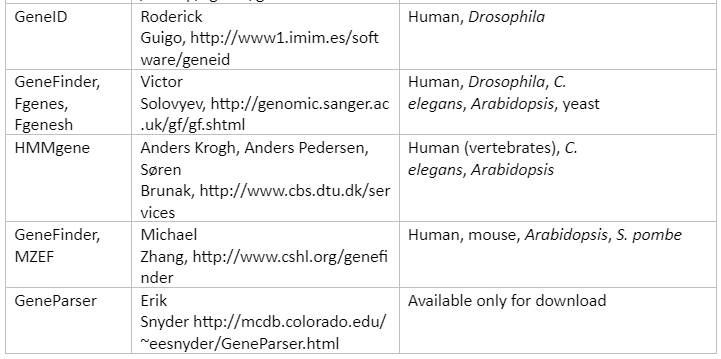
The analysis of DNA sequencing helps us to identify genomic regions with exons, intron and intergenic regions. This information is very important in gene prediction and also allows us to understand the genome at gene level. The tools that can be used for gene prediction are tabulated below:
Table 2: Tools for Gene Prediction

Step Two: Quality check Sequence Alignment
The sequence analysis also allows us to have sequence alignment. This helps us to understand the sequence similarities between our input sequence and the one that is already present in the database. By knowing sequence similarity, we understand which organism or genes are closely related to our specimen of interest. The two popularly known algorithm used for similarity searches are the Smith–Waterman algorithm (local alignment) and the Needleman–Wunsch algorithm (global alignments)
It also helps us to identify single nucleotide polymorphism (SNP), this is the result of variation present at a single position in the genome. SNPs are not only responsible for genetic variation present in a population but also, they are at certain instances associated with a specific disease. Due to this application, the discipline of personalized medicine was developed.
Some of the sequence analysis tools along with their description is given by:
Table 3: Tools for sequence analysis


Molecular Phylogenetics:
Molecular phylogenetics is the study of evolutionary relationships at molecular level. With the help of computational tools and methods, we can determine conserved regions of organisms in their genome and also we can identify marker sequences that can help us to classify them in their respective group or category. For phylogenetic, the common analysis tools are PAUP (phylogenetic analysis using parsimony) and PHYLIP (phylogenetic inference package), etc. Some of the important bioinformatics tools are given by table 4:
Table 4: Important tools for phylogenetics analysis

Transcriptomics
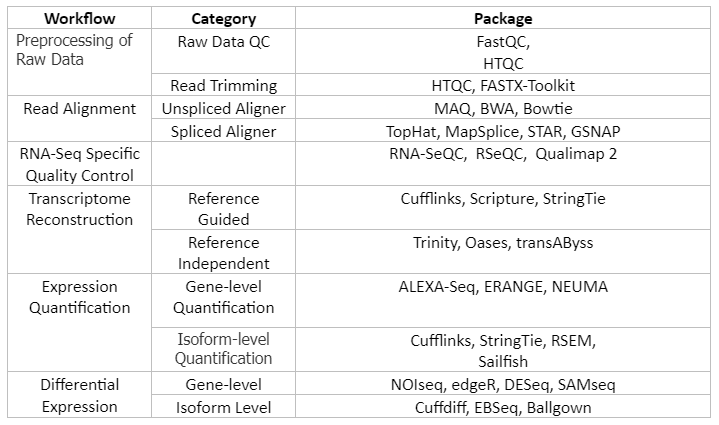
Gene expression analysis is very important in life sciences. Transcriptome data that is obtained from different types of cells help the researchers to deeply understand what constitutes a specific type of cell, how normal cellular functions are performed and what are the changes that can modify the normal functioning of the gene and make it the contributor to disease. Also, when we align the transcriptome of each cell, we can have a complete picture of genome wide gene expression. For transcriptomic studies bioinformatics tools are used to analyze data from microarray or high-throughput sequencing techniques like RNA-sequencing, ChIP-seq etc. The simple workflow and tools that can be used for analysis is given by table 5:
Table 5: Selected list of RNA-seq analysis workflow and programs
Proteomics
Proteome is an entire complement of protein present in an organism. The study of proteome with the help of computational tools is termed as proteomics. The proteomics plays a pivotal role in determination of functions of proteins, presence or absence of components and structural dynamics of proteins.
The bioinformatics analysis of proteomic data obtained from mass spectrometry (MS) is given by folloeing figure 2:
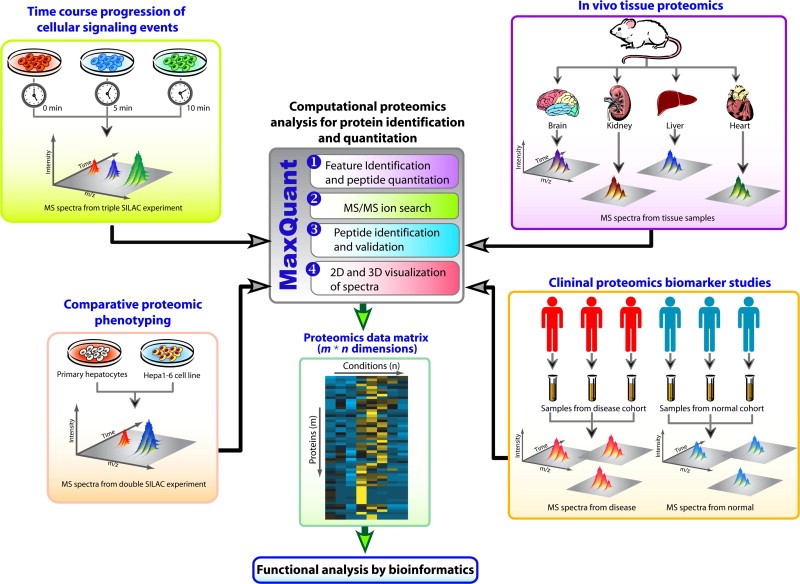
Figure 2: Representation of quantitative data generation in MS of proteomics. The current spectrometric based proteomics combines experimental data, use advanced MS instrumentation to produce multidimensional data set. These datasets are obtained from either isotopic-labeled samples (such as SILAC) (left side of the figure) which are mostly useful to determine temporal pathways of cellular signaling events or in comparative proteomic phenotyping experiments across multiple levels. The right side of the figure show the proteome that are not isotopically labelled like tissue or other clinical samples can also be analyzed through ‘label-free’ approach. The MaxQuant is a computational software which allows analysis of this complex data set and generates a multidimensional data matrix which has a lot of information on protein and peptide. Also for more functional analysis, more bioinformatics tools can be used.
Protein Interaction Networks
Proteins are very important biomolecules; they coordinate many life processes in an organism. The proteins interact with each other and other molecules to accomplish a cellular function. For instance all enzymes are protein in nature and they are taking part in major signaling pathways for cellular respiration and metabolism. In order to understand various processes, researchers are trying to develop protein interaction networks that includes multiprotein complexes, protein trafficking, signaling pathways and protein interactions. Bioinformatics helps in this research by representation, reconstruction and modeling of various protein networks. Also, there are protein database available for retrieval of information. The example of two comprehensive protein database includes Pathguide and BioGRID.
An overview of protein analysis with the help of bioinformatics is given by:
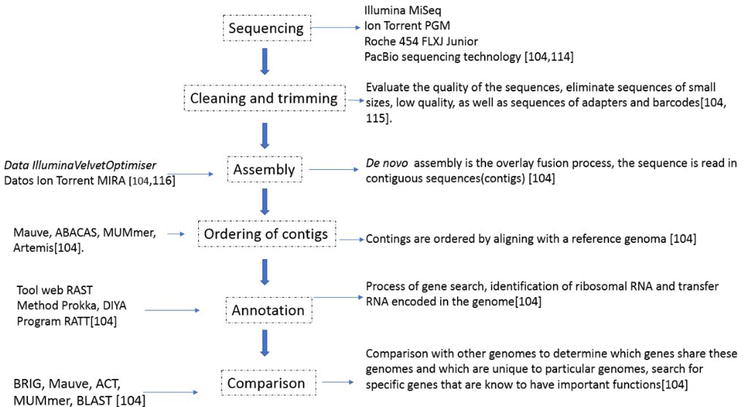
Figure 3: Protein analysis overview
Drug discovery
Bioinformatics analysis has changed the dynamics of drug discovery by providing with an efficient way of identifying potential drug targets. There are variety of bioinformatics tools that allow virtual screening of hundreds and thousands of potential small molecules that can bind with target protein and either activate or inhibit it depending upon its role. In drug discovery, the structure of target protein is quite important. Different types of bioinformatics tools allow 3d structure prediction of proteins. the methods which are generally used for 3d structure prediction of proteins can be classified into two categories:
Comparative modelling (Homology modelling): It is considered to be the accurate method for structure prediction and it is widely used. Homology modelling is based on assumption that all members of a protein family have the same type of fold that is characterized by a core structure, robust enough to overcome modifications. This technique make use of experimentally determined protein structure of template and compare it with sequence of target protein to predict 3d structure.
De novo modelling : It is a computationally intensive way to determine tertiary structure of proteins through amino acid sequence.
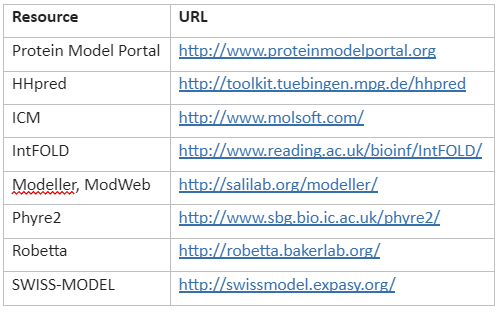
Frequently used tools and servers for homology modelling is given by Table 6:
Table 6: Tools and servers for homology modelling
The next crucial step in computer-aided frug discovery is the virtual screening. For this step, molecular docking (MD) is done. MD has ability to predict the structure of protein-ligand complex and binding affinities between protein and ligand. This information is essential for lead optimization. MD tools includes MEGADOCK, ZDOCK, AutoDock etc.
After the ligands have been identified, they are further optimized and experimentally tested as a potential drug.
Conclusion
The 21st century is all about information technology and its incorporation into life sciences have made analysis easy and efficient. It has led to discovery of new drugs, new protein function and better understanding of the structure and function of the genome. But still, a lot of work needs to be done as most of the computational tools are not user-friendly and require a specific set of skills to operate them. Thus, it is necessary to simplify the computational tools, make them more accurate and efficient.

really appreciate this blog because I like to study Bioinformatics and Molecular Biology
Good information