
Definition
RNA-seq (RNA-sequencing) is a procedure that can analyze the amount and arrangements of RNA in an example utilizing next generation sequencing (NGS). It examines the transcriptome, demonstrating which of the qualities encoded in our DNA are turned on or off and how much.
Why it is used?
RNA sequencing (RNA-Seq) utilizes the abilities of high-throughput sequencing techniques to give understanding into the transcriptome of a cell. Contrasted with past Sanger sequencing-and microarray-based strategies, RNA-Seq gives far higher inclusion and more noteworthy goal of the unique idea of the transcriptome.
Applications
RNA-seq allows us to explore and find the transcriptome, the complete cell content of RNAs including mRNA, rRNA and tRNA. Understanding the transcriptome is vital in case we are to associate the data in our genome with its utilitarian protein articulation. RNA-seq can let us know which qualities are turned on in a cell, what their degree of record is, and at what times they are actuated or closed off.This permits researchers to comprehend the science of a cell all the more profoundly and evaluate changes that might show illness. Probably the most well known procedures that utilization RNA-seq are transcriptional profiling, single nucleotide polymorphism (SNP) identification,RNA altering and differential quality articulation examination.
This can give analysts indispensable data about the capacity of qualities. For instance, the transcriptome can feature every one of the tissues where a quality of obscure capacity is turned on, which may show what its job is. It additionally catches data about elective joining occasions, which produce various records from one single quality succession. These occasions would not be gotten by DNA sequencing. It can likewise distinguish post-transcriptional alterations that happen during mRNA handling.
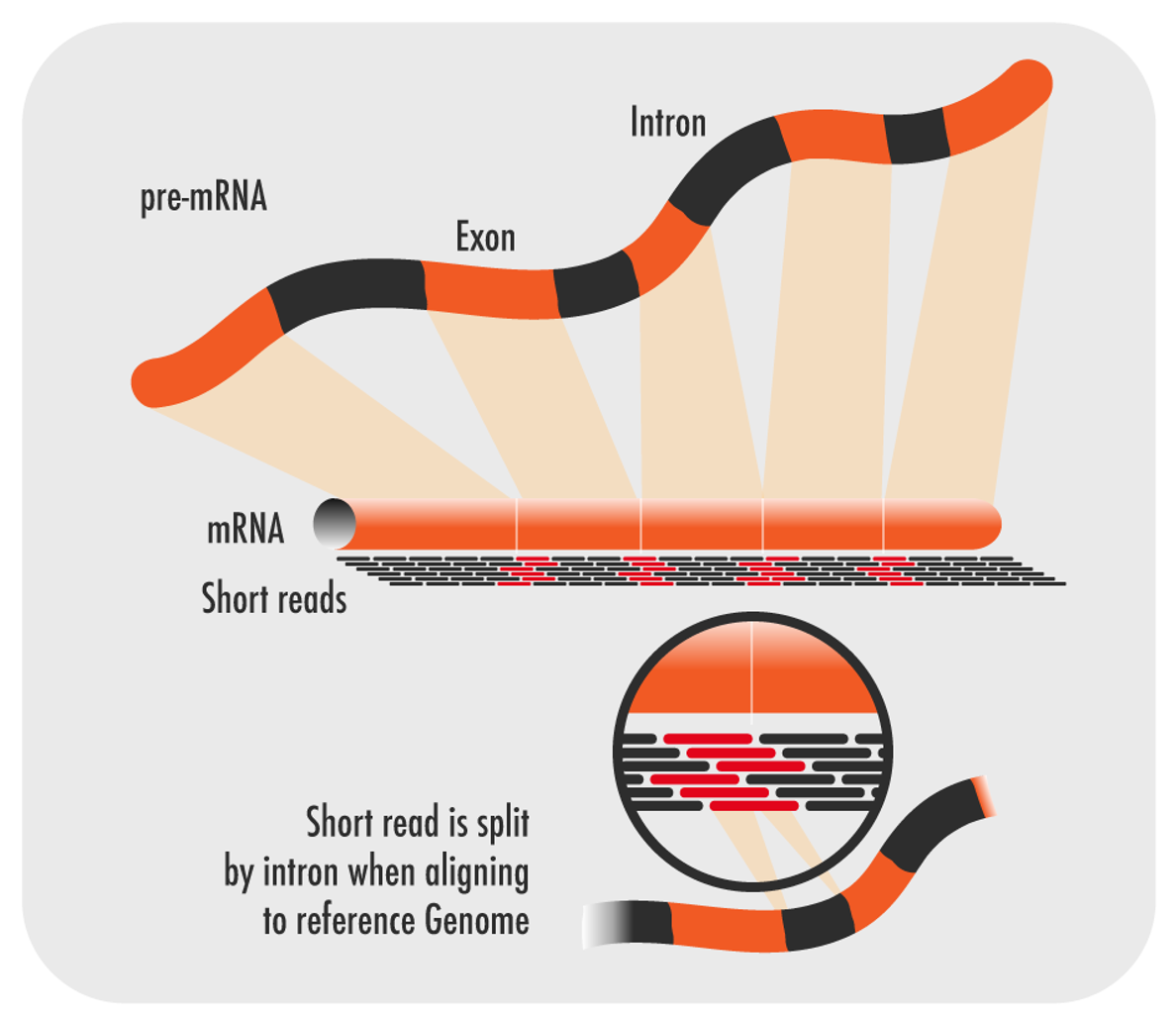
RNA-seq information utilizes short peruses of mRNA which is liberated from intronic non-coding DNA. These peruses should then be adjusted back to the reference genome.
Methods
mRNA Sequencing
Delicately and precisely evaluate quality articulation, recognize known and novel isoforms in the coding transcriptome, distinguish quality combinations, and measure allele-explicit articulation.
Targeted RNA Sequencing
Dissect quality articulation in an engaged arrangement of qualities of interest. Designated RNA-Seq can be accomplished by means of one or the other improvement or amplicon-based methodologies.
Ultra-Low-Input and Single-Cell RNA-Seq
Utilize profound RNA-Seq to analyze the signs and conduct of a cell with regards to its general climate. This strategy is invaluable for scholars concentrating on cycles like separation, multiplication, and tumorigenesis.
RNA Exome Capture Sequencing
Accomplish practical RNA exome investigation utilizing arrangement explicit catch of the coding districts of the transcriptome. Ideal for bad quality examples or restricted beginning material.
Total RNA Sequencing
Precisely measure quality and record bounty and identify both known and novel components in coding and various types of noncoding RNA.
Small RNA Sequencing
Disconnect and succession little RNA species, like microRNA, to comprehend the job of noncoding RNA in quality quieting and posttranscriptional guideline of quality articulation.
Profoundly succession ribosome-ensured mRNA sections to acquire a total perspective on the ribosomes dynamic in a cell at a particular time point, and foresee protein plenitude.
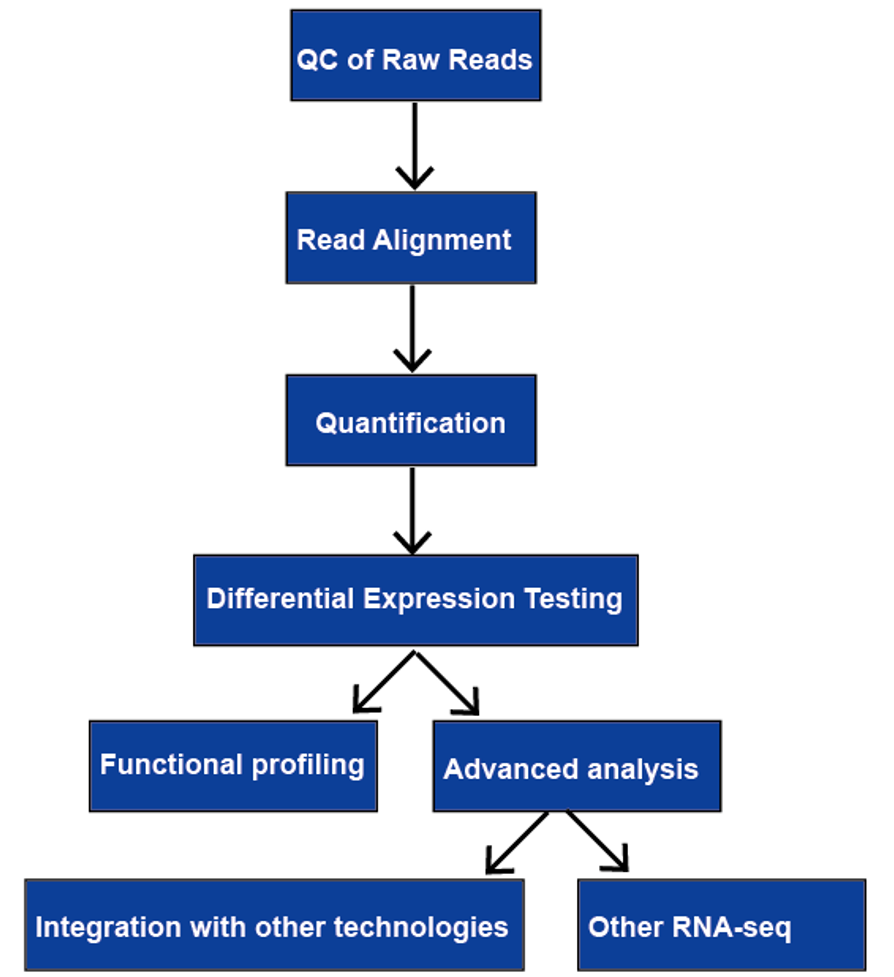
Bioinformatics Workflow of RNA-Seq
RNA-sequencing (RNA-seq) has a wide scope of utilizations, and there is no ideal pipeline for all cases. We audit every one of the significant stages in RNA-seq information examination, including quality control, read arrangement, measurement of quality and record levels, differential quality articulation, useful profiling, and progressed investigation.
Step 1-Quality control of raw reads
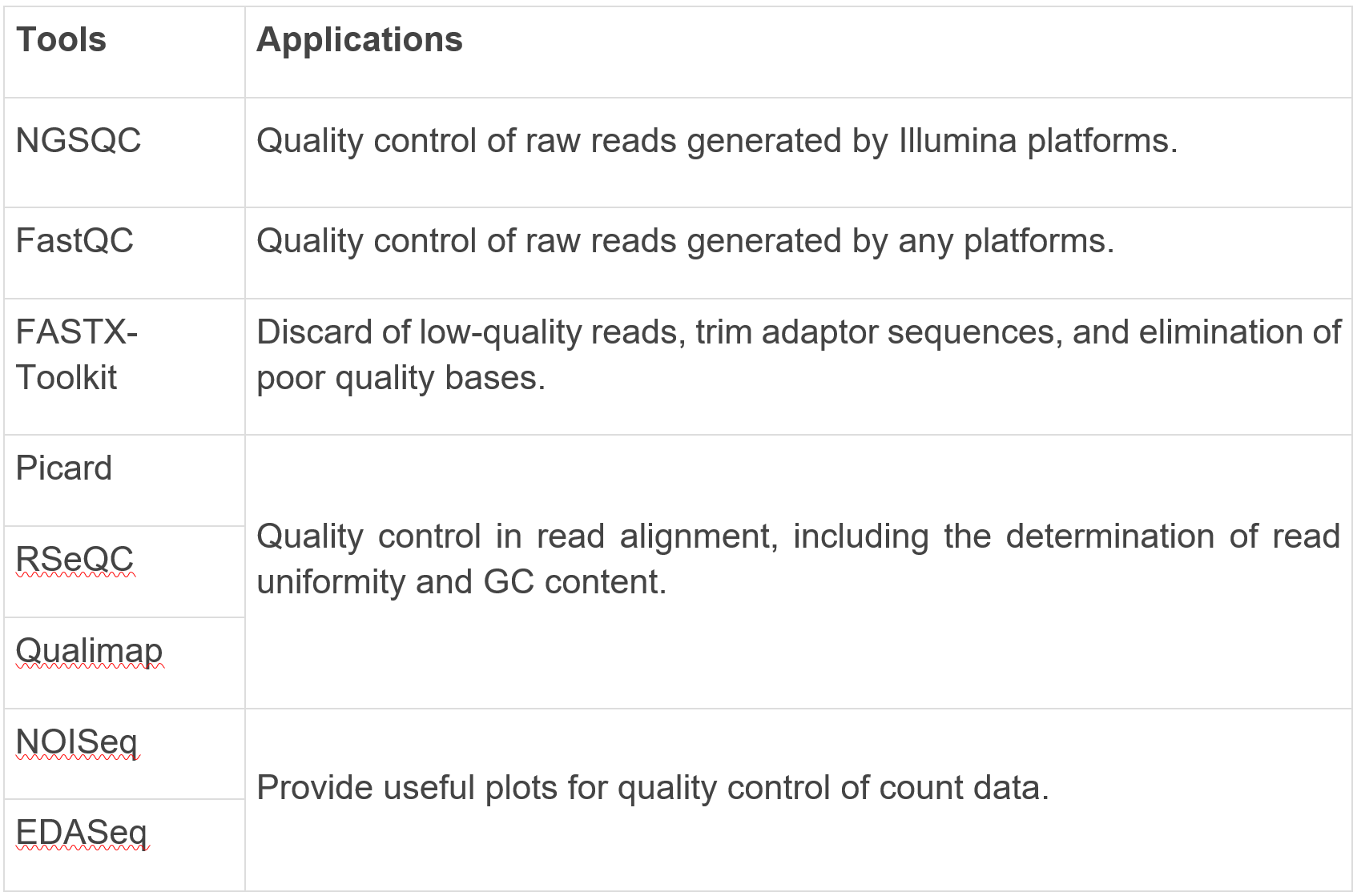
Quality control of RNA-seq raw reads comprises of examination of grouping quality, GC content, connector content, overrepresented k-mers, and copied peruses, committed to identifying sequencing blunders, defilements, and PCR antiques. Peruse quality declines towards the 3′ finish of peruses, bases with bad quality, thusly, they ought to be taken out to further develop mappability. Notwithstanding the nature of crude information, quality control of crude peruses additionally incorporates the investigation of read arrangement (read consistency and GC content), evaluation (3′ inclination, biotypes, and low-counts), and reproducibility (connection, head part examination, and bunch impacts)
Tools of quality control
Step 2-Read alignment
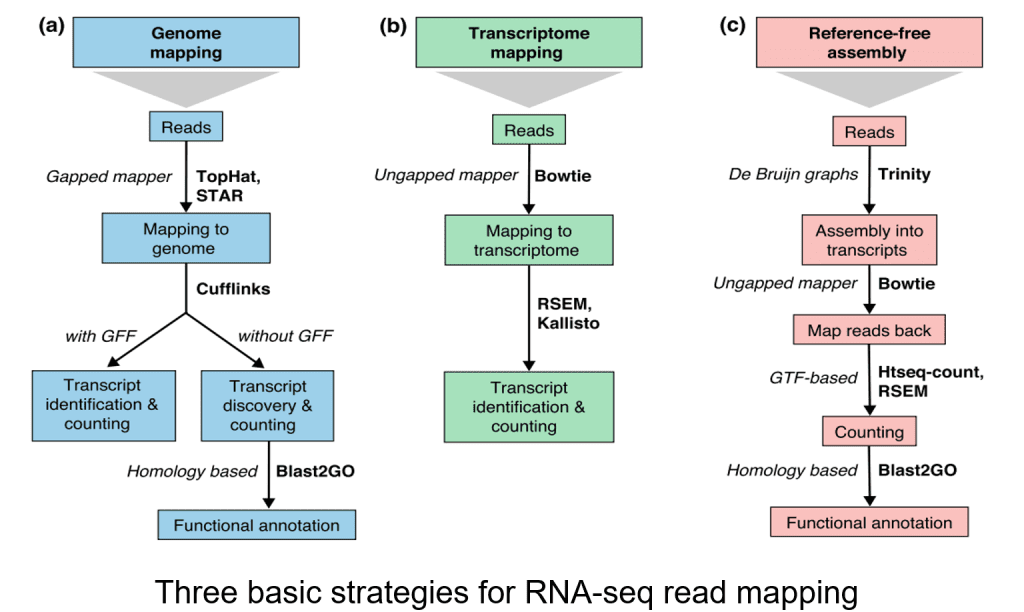
There are by and large three systems for read arrangement, genome planning, transcriptome planning, and again get together. Whether or not a genome or transcriptome reference is accessible, peruses may plan remarkably or be appointed to different situations in the reference, which are alluded to as multi-planned peruses or multireads. Genomic multireads are for the most part because of dull arrangements or shared spaces of paralogous qualities. Transcriptome multi-planning emerges all the more frequently because of quality isoforms. Thusly, record distinguishing proof and measurement are significant difficulties for then again communicated qualities. At the point when a reference isn’t free, RNA-seq peruses are collected once more utilizing bundles like SOAPdenovo-Trans, Oases, Trans-ABySS, or Trinity. PE strand-explicit and long-length peruses are liked since they are more educational.
Step 3 Transcript quantification
Transcript quantification can be used to estimate gene and transcript expression levels.

Differential expression testing
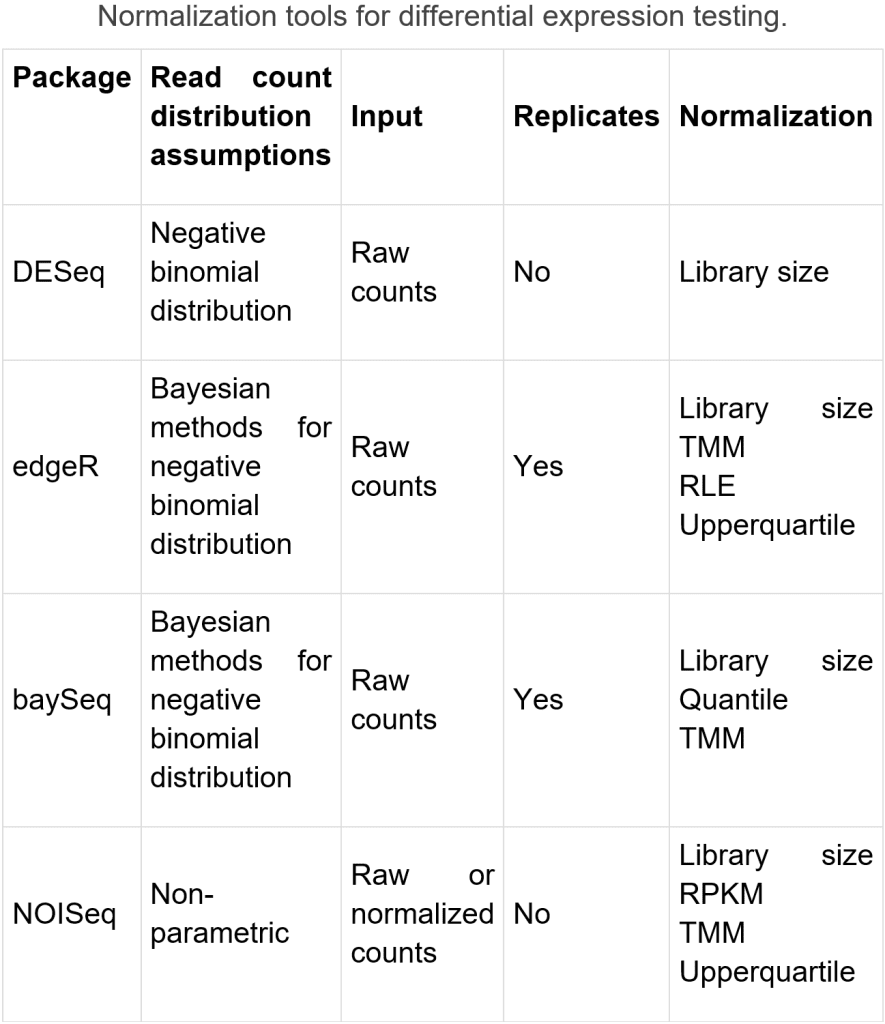
Differential expression testing is utilized to assess on the off chance that one quality is differentially communicated in one condition contrasted with the other(s). Normalizing strategies should be taken on prior to looking at changed examples. RPKM and TPM standardize away the main factor, sequencing profundity. TMM, DESeq, and UpperQuartile can overlook profoundly factor and additionally exceptionally communicated highlights. Different components that meddle with intra-test correlations include record length, positional predispositions in inclusion, normal piece size, and GC content, which can be standardized by apparatuses, like DESeq, edgeR, baySeq, and NOISeq. Cluster impacts might in any case be available after standardization, which can be limited by fitting exploratory plan, or eliminated by strategies like COMBAT or ARSyN.
Alternative splicing analysis
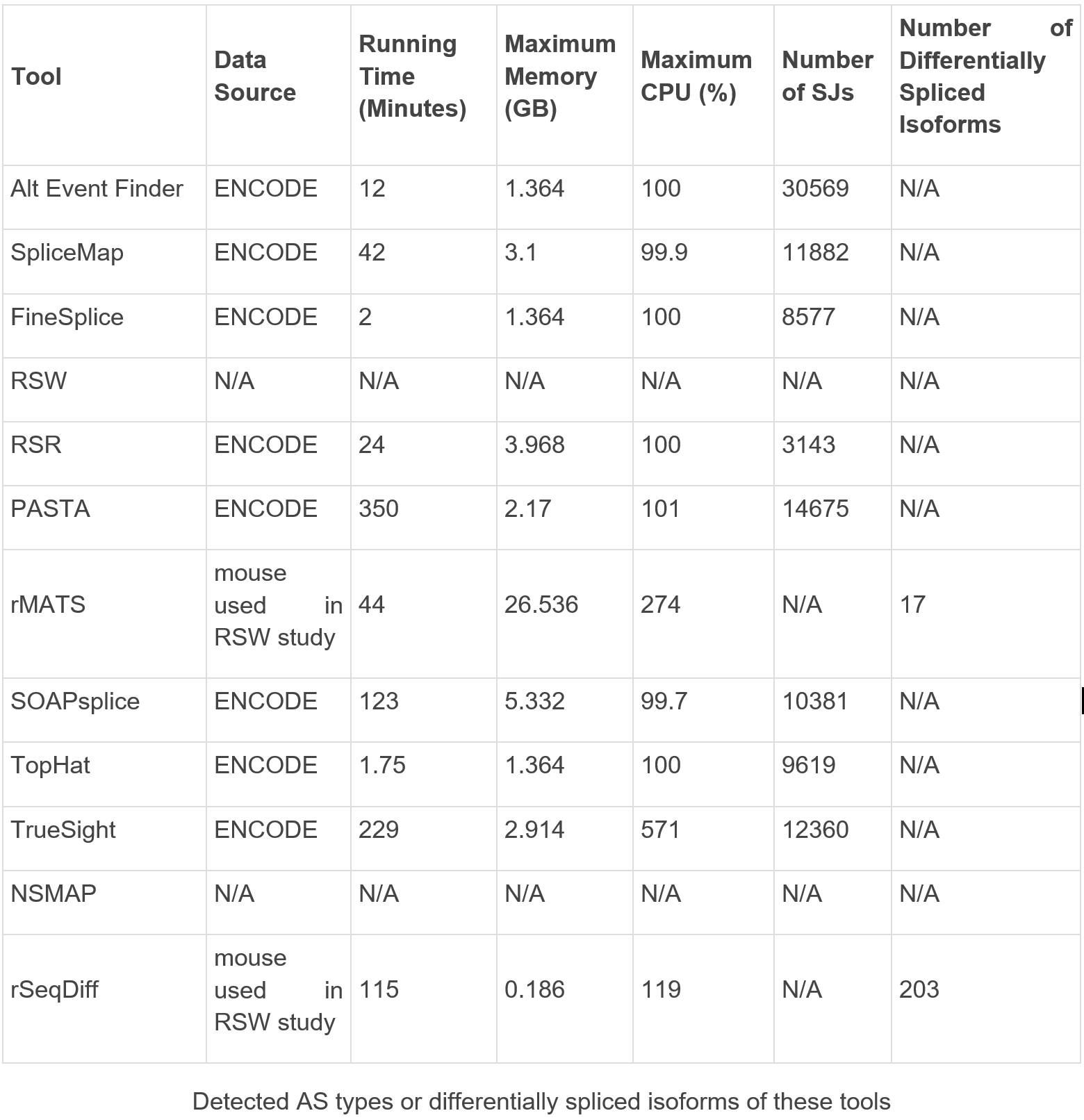
Elective joining (AS) is a posttranscriptional cycle which creates various records from a similar quality and is indispensable in light of natural improvements by delivering different protein items. Various bioinformatics instruments have been created to identify AS from trial information. The correlation of these location instruments utilizing RNA-seq information was directed by Ding in 2017, and the outcomes are displayed in Table 7. They have shown that TopHat and its downstream device, FineSplice, are the quickest instruments, while PASTA is the slowest program. Moreover, AltEventFinder can identify the biggest number of intersections, and RSR recognizes the most reduced number of intersections. Different apparatuses, like TopHat, are probably going to identify bogus positive ones.
Visualization
There are numerous bioinformatics devices for the perception of RNA-seq information, including genome programs, like ReadXplorer, UCSC program, Integrative Genomics Viewer (IGV), Genome Maps, Savant, devices explicitly intended for RNA-seq information, like RNAseqViewer, just as certain bundles for differential quality articulation examination that empower the representation, like DESeq2 and DEXseq in Bioconductor. Bundles, for example, CummeRbund and Sashimi plots, have additionally been created for representation select purposes.
Functional Profiling
The most recent advance in a standard transcriptomics study is by and large the portrayal of the atomic capacities or pathways in which differentially communicated qualities are involved. Quality Ontology, Bioconductor, DAVID, or Babelomics contain explanation information for most model species, which can be utilized for practical comment. Concerning novel records, protein-coding records can be practically explained utilizing orthology with the assistance of information bases like SwissProt, Pfam, and InterPro. Quality Ontology (GO) considers some exchangeability of utilitarian data across orthologs. Blast2GO is a well known instrument that permits monstrous explanation of complete transcriptome against an assortment of data sets and controlled vocabularies. The Rfam data set contains most all around described RNA families that can be utilized for useful comment of long non-coding RNAs.
Advanced analysis
The advanced analysis of RNA-seq usually includes other RNA-seq and integration with other technologies. More information on applications of RNA-seq,
Benefits of RNA Sequencing
- Broader dynamic reach empowers more touchy and precise estimation of quality articulation
- Not restricted by earlier information – catches both known and novel elements.
- Can be applied to any species, regardless of whether reference sequencing isn’t accessible.
- A better worth, frequently conveying benefits at a tantamount or lower cost per test than many clusters.
