
What Is Cancer Genomics?
The 21st century is the era of technology and innovation. With advancements in techniques and the development of high throughput sequencing like Next-generation sequencing (NGS) and various computational analysis, genetics and molecular biology are transformed. Cancer is also a genetic disease that occurs due to changes in the genome of the cells which results in uncontrolled cell division.
Cancer is defined as a disease that is caused due to uncontrolled cell division resulting in producing a large number of undifferentiated cells that spread to other parts of the body. Cancer cells can form at any stage of life and in any part of the body. There can be many reasons for cancer cells among which the mutations caused by natural causes (faulty cell division or DNA replication) and unnatural sources (radiations). Currently, with the development of high-throughput sequencing techniques, a lot of research is being carried out on cancer. Thus, cancer genomics is the discipline that studies the entire DNA sequence and gene expression between normal host cells and tumor cells. The main aim is to understand the underlying genetic basis of tumor cell metastasis (proliferation) and cancer genome evolution under mutation and selection by the immune system, body environment, and therapeutic intervention.
Why are omics in Cancer a necessity?
The utilization of NGS to characterize tumors of human beings has enabled us to understand the biological origin of distinct types of cancer cells which will be helpful in the development of targeted therapies. The genomics in cancer also helps us to identify and discover biomarkers associated with drug response and resistance thus, allowing us to make a relevant decision regarding the treatment of patients. The NGS also reveals:
Small Indels i.e., insertion or deletions
Mutation in sequences
Copy number variations
Absence of heterozygosity in tumor DNA samples
In addition to this, sequencing of RNA derived from tumors allows identifying differential expression of genes, small RNAs, gene fusion, allele-specific expression patterns, and aberrantly spliced isoforms. All of the above-mentioned advantages make cancer genomics an important discipline.
Overview of applications of genomics in cancer
Cancer and Genomics
The current era of genomics commenced in the late 1970s when Frederick Sanger pioneered a DNA sequencing technology that determined the nucleotide sequence of DNA in the genome. After sanger sequencing, efforts were being made to decipher the genetic code which resulted in improvements in sequencing and the establishment of a reference human genome in the 21st century. This reference genome served as the standard to which the genome of other individuals was aligned and compared, it led to annotation of the genome, the discovery of biomarkers, single nucleotide polymorphism, and the link of structural variants to phenotype.
The insight of the reference genome proved quite useful in cancer biology, as we know that cancer development and progression is linked with DNA mutations, so by using sequencing these mutations can be identified which can then have clinical applications. The first cancer-specific mutation was discovered by using sequencing was in HRAS which was present in the T24 bladder cancer cell line. This discovery was proof that sequencing technologies are essential in cancer biology. There are three generations of sequencing technologies, their role in cancer studies and their pros and cons are given by following table 1:
Summary of sequencing technologies and Cancer
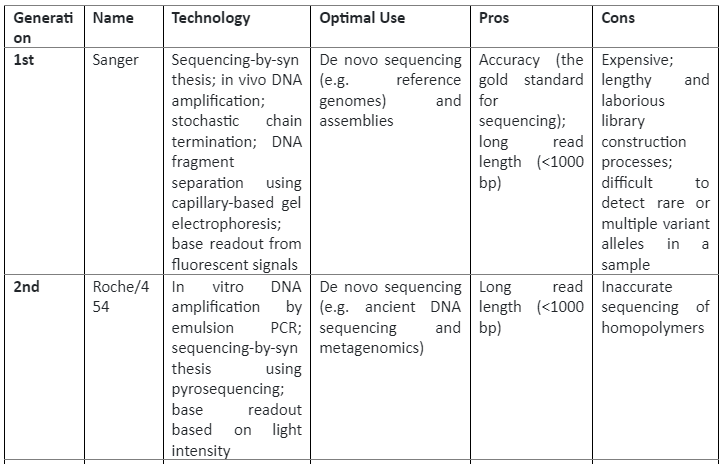
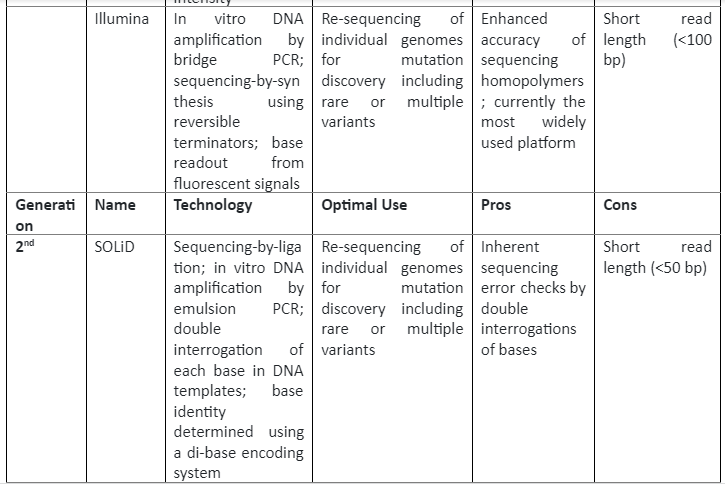
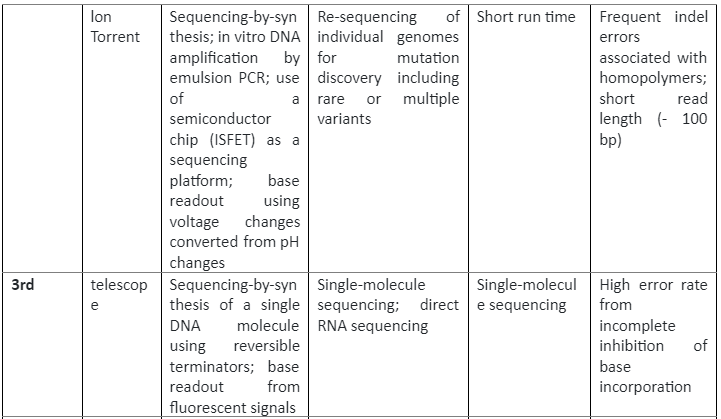
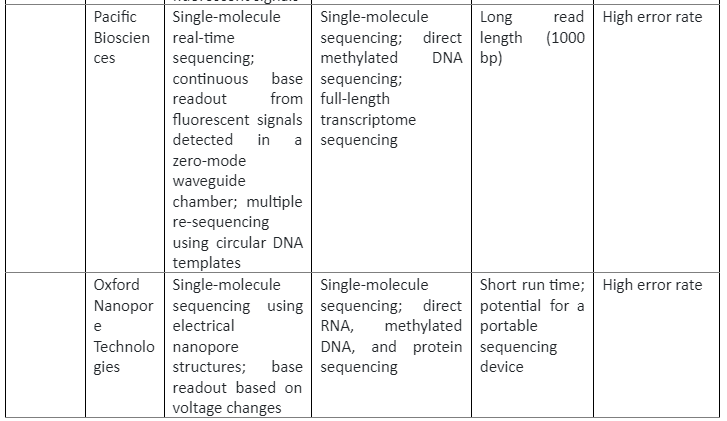
Clinical utility of cancer genomics
After the human genome project was completed in 2001, it was established what the normal genome of a healthy person looks like. After a few years, a catalog was established named “The Cancer Genome Atlas” and the “International Cancer Genome Consortium”. The major aim of these projects was to identify alterations present in different cancer cell types by harnessing NGS. After a brief time, a surge of analytical and genomic data takes place making this field pivotal in cancer research and clinical oncology. Some of the applications are explained below:
Identifying therapeutic targets.
The sequencing and computational analysis have resulted in the identification of new genomic abnormalities involved in tumor development and progression. To inhibit these abnormal mutations, molecular target therapies can be used.
To identify a target for therapy, the genetic changes are identified at the site of tumor growth and change. Then, a protein that is associated with tumor growth and its absence in healthy cells is determined as a target. After the target is identified then, specific molecules are small molecules are screened which interact with that protein and inhibit it. An example of this precision oncology is the trastuzumab which is used to treat patients with HER2-amplified metastatic breast cancer. HER2 is a transmembrane tyrosine receptor that is overexpressed to almost 25-30% in breast cancer cases. It is unique from all the other receptors of its family by the fact that it has no ligand. HER2 was considered an ideal target for cancer drug treatment due to:
HER2 levels are strongly proportional to the level of carcinogenesis as demonstrated through loss or gain of function experiments.
HER2 overexpression is found to be associated with both the primary tumor and metastatic sites, making it an effective drug target.
The level of HER2 is quite high in a cancer cell in comparison to healthy cells, thus making it a potential target without causing less effect on normal cells.
After screening and trials trastuzumab was found to be the inhibitor of HER2. At the moment, many FDA-approved genotype-directed therapies are being used.
Complexity aspects of cancer
There are diverse and very fast-evolving mutations that are clinically relevant. This means that to have a comprehensive analysis, the molecular profiling platforms must have the potential to incorporate all types and classes of genomic alterations. For this purpose, hotspot panels are mostly used that detect the recurrent mutation in EGFR, BRAF, and many other genes. No doubt that this method is broader but hybridization-capture-based panels are considered better for the detection of deletions, amplification, and chromosomal rearrangements. Among these rearrangements, one that results in the production of kinase gene fusions is very important. Previously it was thought that these fusions were exclusively associated with hematological malignancies (like BCR-ABL mutations in CML patients) but now they are found associated with many solid tumors. For example, the presence of fusion containing RET, ALK, or ROS 1 in patients having Non-Small Cell Lung Cancer (NSCLS). This evidence has underscored the need for new techniques that enable us to find out about all the therapeutically actionable fusions.
Evolution of clinical trial
The novel clinical trial was developed after similar mutations were identified across many cancerous cell types. These novel clinical trials were needed to detect the efficacy of the drug. For instance, in one of the trials known as the basket trials, patients were recruited based on the specific type of gene alternation rather than the type of tumor. In basket trials, molecularly targeted drugs were used which were best suited for low-frequency mutations taking place across many different types of cancer. Thus, in this way, individuals were able to be treated more robustly and responsively in comparison with studies focused only on the overall disease. Also, these therapies have increased access to alternation-specific therapy, especially in patients having rarer types of cancers.
Acquired resistance challenges
Although, the best application of cancer genomics is mutation-specific therapy, and no doubt these therapies result in a remarkable initial response but there are chances of the presence of other mutations in conjunction with the one being treated. In addition to this, biological factors also can cause innate drug resistance in some patients. This drug resistance is more common in patients having metastatic cancers after prolonged treatment with targeted therapy.
Besides, there are certain secondary mutations present in the target that can make it unresponsive to inhibition and also have the ability to activate many downstream effectors or even bypass them to initiate tumor reactivation even in the presence of the drug. For instance, secondary mutations in CML patients having BCR-ABL mutation have been identified and this has resulted in the development of more potent inhibitors known as the second and third-generation inhibitors.
Genomics has discovered many new mechanisms in tumor biology which are involved in acquired resistance. The genome-based analysis for this purpose can be divided into two main approaches:
In the first method, a comparison of sequence data is done among the cohort of patients having pretreated metastatic cancer with the patients having untreated primary tumors. Mutations will be identified in patients having metastatic disease. For example, identification of ligand-binding domain mutation in gene encoding estrogen receptor (ESR1) due to their high frequency in patients having metastatic breast cancer.
In the second approach, the comparison of mutational profiles by sequencing paired pretreatment and post-treatment biopsy samples of the same patients at different times can also help in the identification of novel acquired mutations. For instance, paired sequencing analysis of BRAF-mutant melanoma biopsy samples pre and post-treatment with RAF inhibitor has elucidated acquired mutations in the MAPK signaling pathway and MEK 1.
Implementation Challenges
No doubt, there is a surge of target-specific cancer therapies but keeping track of all the mutations, drug response, acquired mutations and many other factors make it very difficult for clinical practitioners and oncologists to keep track of clinical implications. Many academic cancer centers have specific tumor boards and they make treatment recommendations based on genomics and clinical outlook. But, if we see this from a broader perspective, it is not an easy decision-making process as the number of cancer patients is increasing day by day. Also, even a single type of tumor has distinct features, functions and has mutations at different regions of the same gene and even in some cases, different cancer cells have the same mutations. Several groups are now making efforts to take information and knowledge from expert recommendations, scientific literature, and clinical guidelines and form databases to support clinicians. This will allow them to make a better decision and recommend an optimal treatment.
In addition to this, despite the precision of oncology approaches, there are only a few subsets of patients that have a specific type of actionable mutations. Only for such type of mutations, do the clinical data exists and therapy is then recommended. There is another type of recurrent mutation which is put in the category of “undruggable” mutations and other actionable targets are not characterized yet. Also, “actionable” is defined and assessed differently across many institutions and studies. In addition to this, there is an only minority of cancer patients are enrolled in clinical trial cohorts which have actionable mutations detected with the help of clinical sequencing. This is the reflection of limitations in trials and awareness among both patients and physicians.
Tumor mutational profiling is not limited to the identification of actionable mutations but also can predict response to targeted therapies. Complex genomic features and specific mutational signatures presence can be used to make informed clinical decisions. Also, tumor mutational load has appeared as the biomarker that can be correlated with clinical advantage from immune-checkpoint inhibitors. A mutational signature like microsatellite instability (MSI) has been identified as the interpreter of immune checkpoint inhibition response, this results in FDA approving a drug pembrolizumab which has anti-programmed cell death protein 1 (PD-1- antibody to be used in patients having high MSI solid tumors. This was the first-ever approval of a cancer therapy that was only based on genomic biomarkers, irrespective of tissue of origin.
Furthermore, tumor sequencing also provides us with information that is crucial for prognosis or diagnosis and also elucidation of mutations that show a lack of response to particular interventions, reveal occult germline alleles with cancer susceptibility, establish a clonal association of different lesions, and monitoring of disease after treatment. Thus, tumor genome profiling has become the backbone of precision oncology.
Challenges in cancer genomics
The major challenge faced in cancer genomics is the algorithmic analysis of NGS data from multiple sources as every class of alteration needs a distinct computational approach for detection. Also, the level of detection sensitivity is affected by intra-tumoral heterogeneity and copy number alterations. All the approaches of NGS that are used in cancer diagnostic required high depth sequence coverage to detect mutations even in a small fraction of cancer cells. Some of the important challenges in cancer genomics are explained below:
Sample-related Challenges:
The sample-based challenges like the quality and quality of the tumor tissue sample can pose great challenges to genomic characterization through NGS. Tumor samples provide only a small amount of the genetic material especially when they are collected in the form of biopsy samples or fine-needle aspirants. Also, these samples have a very low amount of tumor cellularity due to the presence of non-malignant cells that can cause loss of signal from somatic mutations and hence a low level of sensitivity detection. Various methodologies have been developed to preserve the tissues and maintain the integrity of DNA in samples like the storage of tumor cells as formalin-fixed, paraffin-embedded (FFPE) tissue blocks. But FFPE process can also result in DNA modification and fragmentation.
As the advances in technology have now enabled us to make use of sample preparation methodologies where only small input of DNA samples is required. Due to this, high-throughput NGS sequencing technologies provide us with highly reproducible and robust data. But still, the quality of DNA must be ensured even in small quantities.
NGS-related Challenges:
NGS technologies enable us to do whole-exome, whole-genome, and whole transcriptomic sequencing which elucidate cancer mutations and alternations in cells. But some of the challenges faced in the adaptation of these approaches are:
Use of higher computational requirements
Higher costs than the alternative techniques
interpretability of increasingly complex results in the clinical context
development of multiple types of algorithms or computational tools that can enable us to identify the specific type of mutation in cells.
Future Perspective
No doubt NGS technologies have made us identify genome-specific mutations and the association of germline mutation with cancer but still a great deal of work needs to be done to overcome the challenges faced in cancer genomics today.
