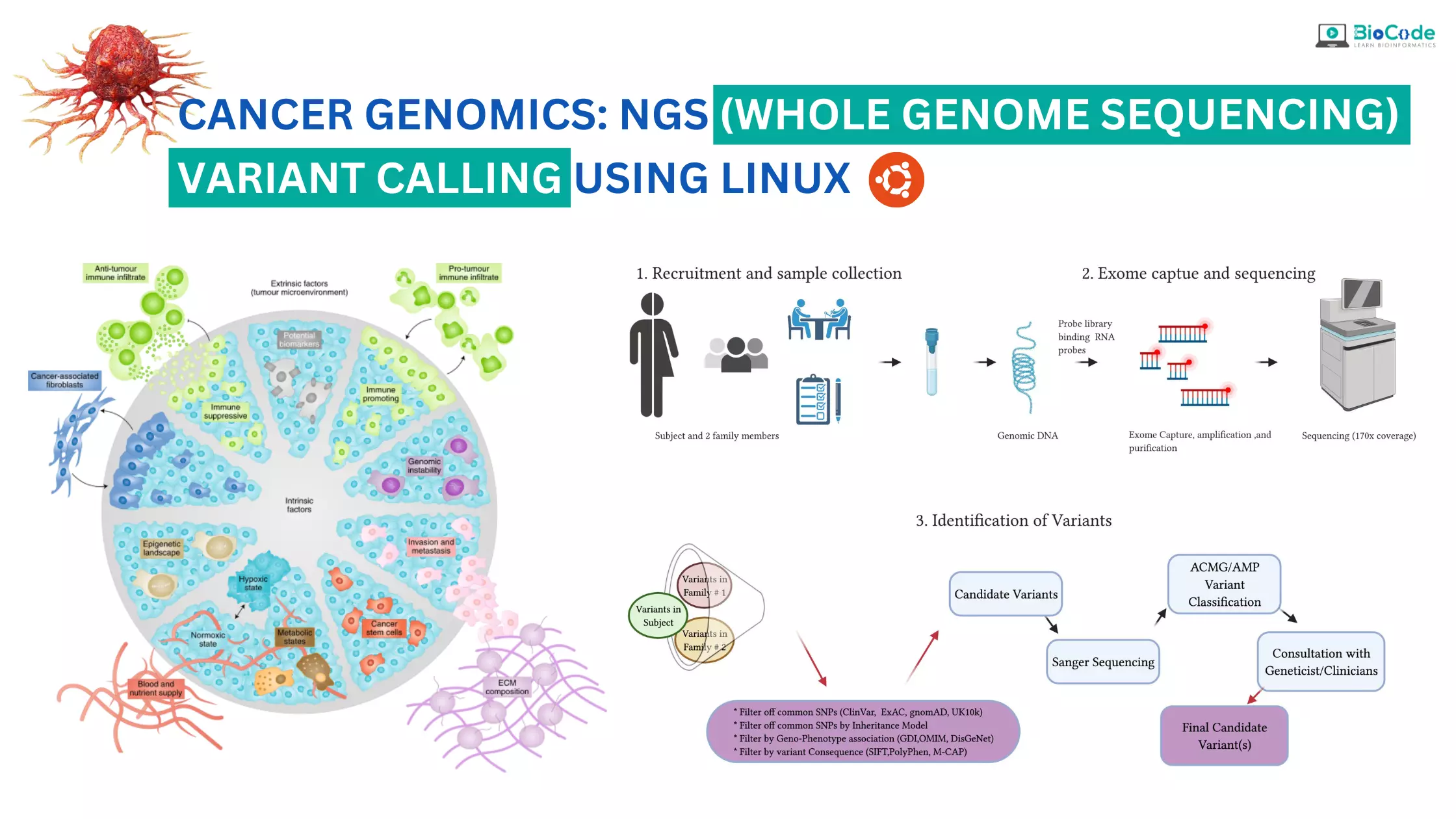
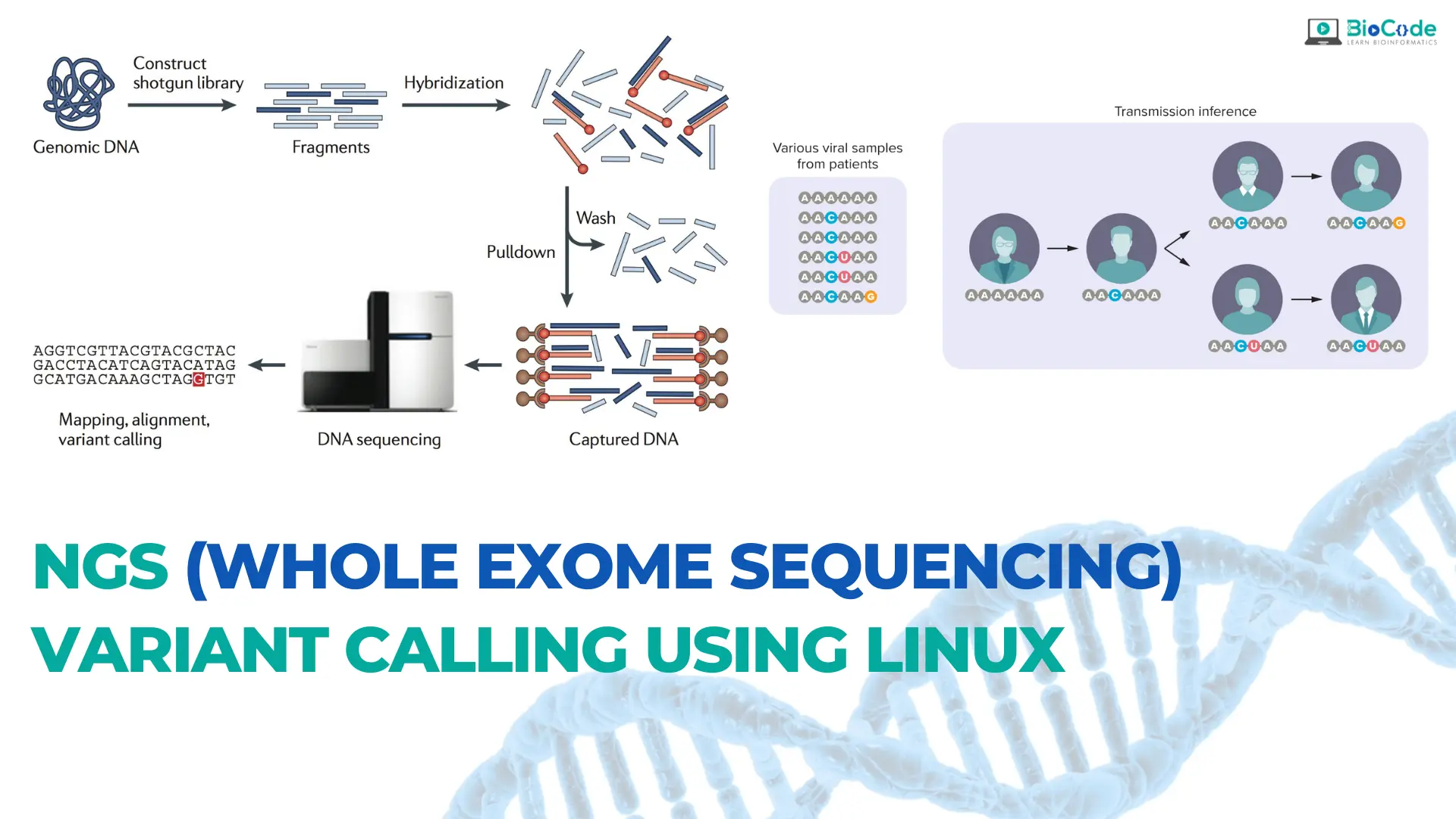
Cancer Genomics: NGS (Whole Exome Sequencing) Variant Calling Using Linux
About Course
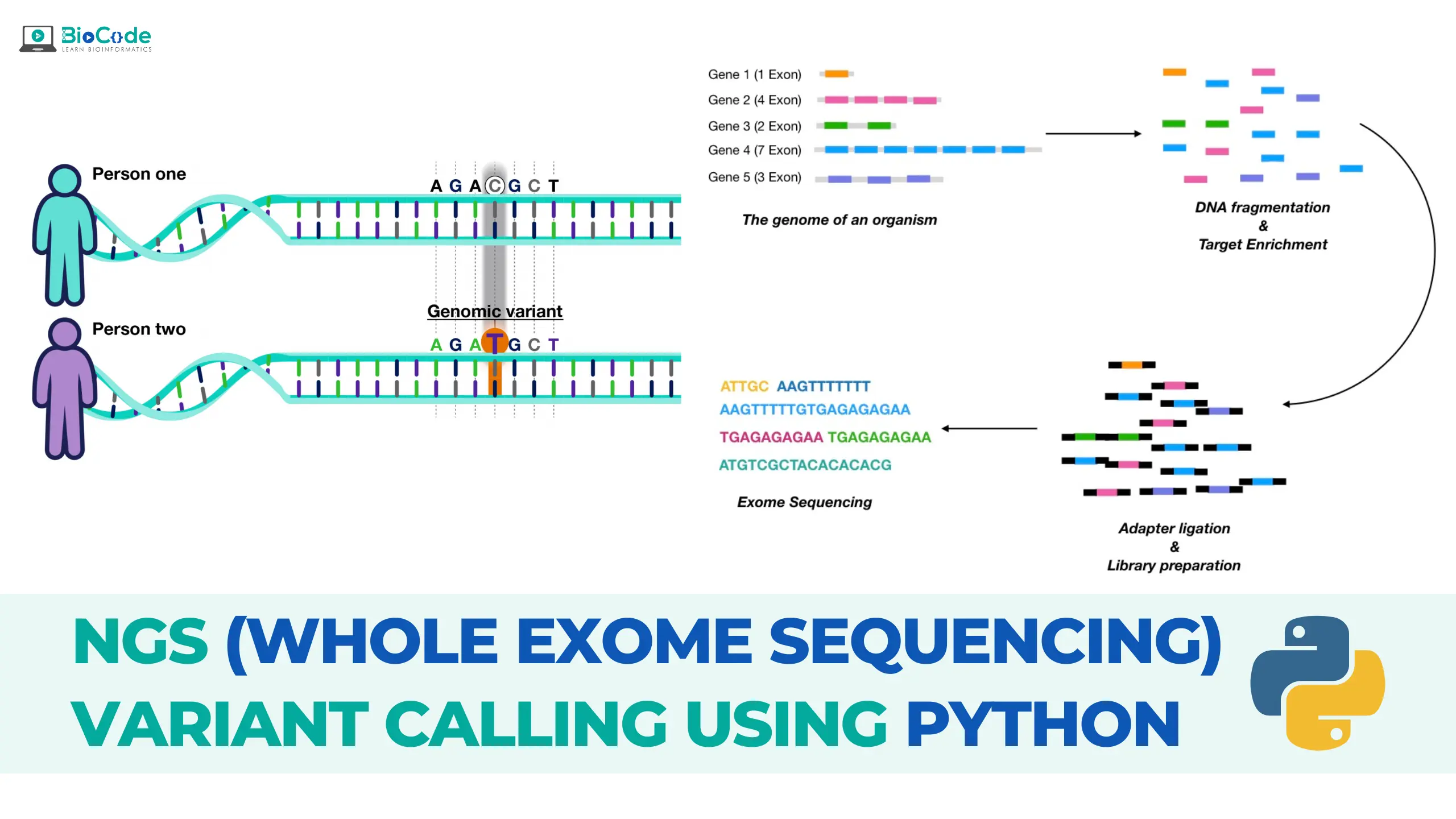
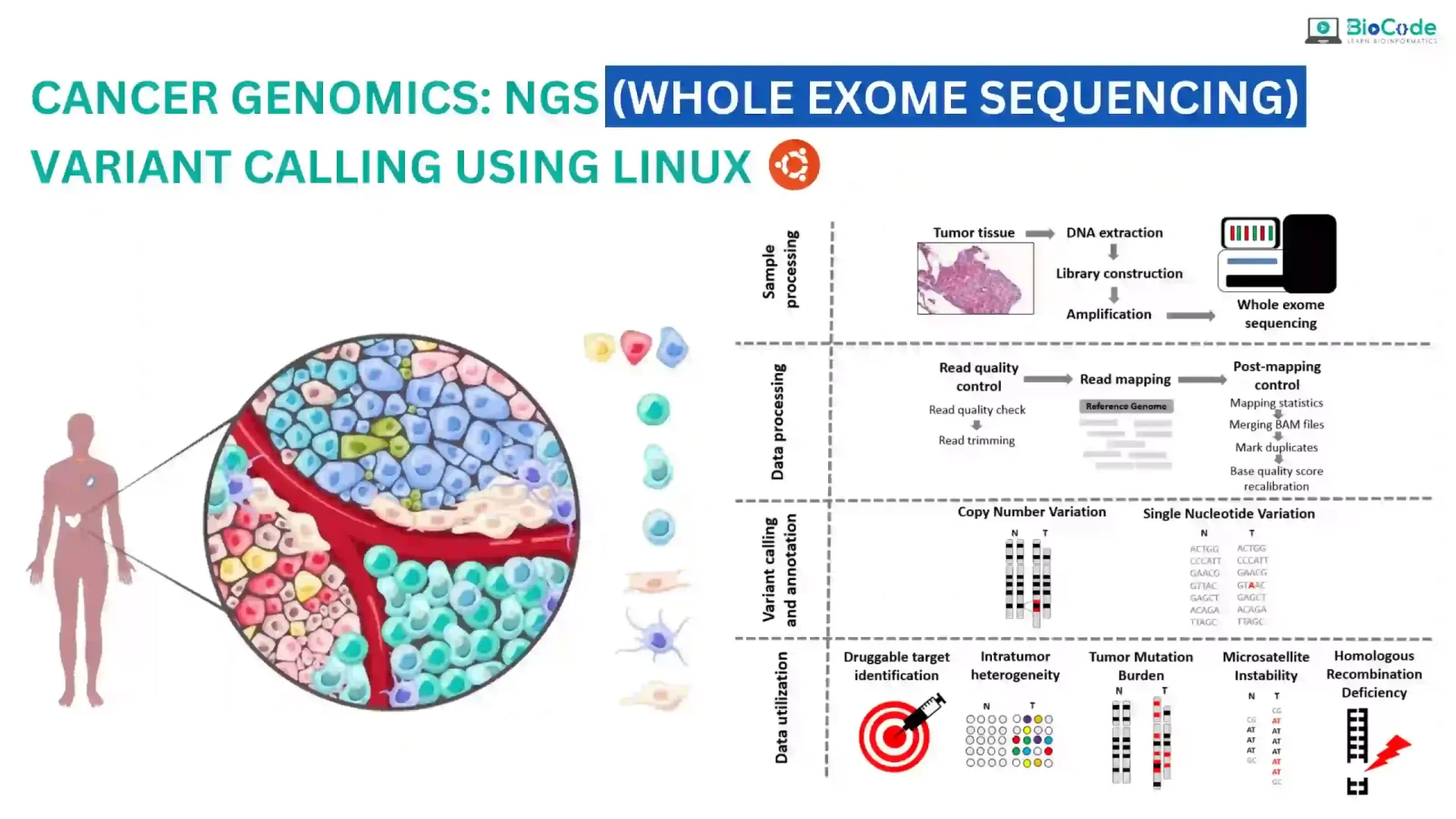
Cancer genomics is a rapidly evolving field that utilizes next-generation sequencing (NGS) technologies to better understand the genetic basis of cancer. One of the most powerful tools in cancer genomics is whole exome sequencing, which enables the sequencing of all protein-coding genes in a genome. However, analyzing the massive amounts of data generated by NGS requires sophisticated computational tools, such as the Linux operating system, which provides a highly flexible and customizable environment for bioinformatics analysis.
BioCode is offering Cancer Genomics: NGS (Whole Exome Sequencing) Variant Calling Using Linux course which is designed to provide students with the knowledge and skills necessary to perform whole exome sequencing (WES) variant calling using Linux. Whole exome sequencing is a powerful technique that can be used to identify genetic mutations and variations that contribute to the development and progression of cancer. Variant calling is an essential step in the analysis of WES data, which involves identifying genetic variants that are different from the reference genome. The course will consist of both lectures and hands-on practical sessions. The course will cover the entire process of NGS variant calling, from initial data pre-processing and alignment to the identification and annotation of genetic variants.
This course is designed to provide a comprehensive understanding of cancer genomics and how NGS technology is used to identify cancer-related genetic mutations. The course is primarily focused on the analysis of cancer datasets using the Linux command line, which is an essential tool in the field of bioinformatics. Students will have the opportunity to work with real-world cancer genomics datasets and gain a deep understanding of the bioinformatics pipelines used in cancer genomics research.
This course is divided into three categories covering an in-depth introduction to NGS and variant calling, hands-on whole-exome variant calling, and additional downstream analysis. Students will gain hands-on experience using bioinformatics tools such as SAMtools and GATK to perform variant calling on NGS data. They will also learn how to visualize and interpret the results of their analyses using popular tools such as IGV and UCSC Genome Browser. No prior knowledge or experience to enroll in this course.
Throughout the course, students will learn how to analyze NGS data for identifying genetic mutations and variations, including substitutions (SNPs) and insertions/deletions (indels) in a raw dataset. Students will learn how to obtain raw whole exome datasets for any organism or disease, perform quality control, trimming, mapping against a reference genome, post-alignment quality control, recalibration of the reads, and finally, how to identify variants in the disease/patient samples to obtain VCF files. Students will also learn how to use various tools to filter and annotate these VCF files, ensuring the accuracy and reliability of their results in the context of cancer genomics research. The course will focus on the identification and analysis of genetic mutations and variations specific to cancer, including somatic mutations and driver mutations.
By the end of this course, students will gain hands-on experience working with real NGS data, specifically in the context of cancer genomics research. They will learn how to troubleshoot common issues and optimize their analysis pipeline for different types of cancer datasets. Students will have a thorough understanding of NGS and whole exome data analysis and will be well-equipped to apply their skills to cancer genomics research and other related fields.
This course will include the following sections:
Section 1: In-depth Introduction to NGS and Whole-Exome Sequencing for Cancer Genomics
Description: Students will be introduced to the fundamentals of NGS, variant calling, and the role of whole-exome sequencing in cancer research. Students will learn about haploid vs. diploid organisms, ploidy and its significance in disease research, and germline vs. somatic mutations. Students will also be introduced to single nucleotide polymorphisms (SNPs) and their types, as well as large-scale variations in genomes and the role of copy number variations (CNVs) in diseases.
Learning Outcomes: Upon completion of this section, students will be able to:
- Describe Next-Generation Sequencing and Variant Calling.
- Explain the Fundamentals of Whole Exome Sequencing.
- Discuss Cancer Genomics and the Role of WES in Cancer Research.
- Explain Haploid vs. Diploid Organisms for Variant Calling.
- Describe Ploidy and Its Significance in Disease Research.
- Describe a Bioinformatics Approach Towards Germline vs. Somatic Mutations.
- Explain Single Nucleotide Polymorphisms and Their Types.
- Discuss Structural Variations.
- Explain CNVs and the role of CNVs in Diseases.
Section 2: Hands-on Whole-Exome Variant Calling
Description: Students will gain hands-on experience with the GATK pipeline for variant calling. They will learn how to retrieve raw whole-exome cancer data, perform quality control, and filter low-quality reads. Students will also learn how to map reads against a reference genome, using multiple tools such as Freebayes, and Mutect2 for variant calling, and filter out low-quality variants using VCFtools. Finally, students will learn how to perform variant effect prediction using various tools such as VEP to identify novel pathogenic variants. Students will also learn how to identify and analyze genetic mutations and variations specific to cancer. They will learn how to identify germline and somatic variants involved in the diagnosis and prognosis of cancer.
Learning Outcomes: Upon completion of this section, students will be able to:
- Explain GATK Pipeline for Variant Calling.
- Retrieve Raw WES Cancer Data.
- Perform Quality Control.
- Trim Out the Bad Quality Reads.
- Perform Mapping of Reads Against a Reference Genome.
- Perform Alignment of Trimmed Reads Against a Reference Genome Using BWA/Bowtie2.
- Perform Post-Alignment Processing of Aligned (SAM/BAM) Files.
- Perform Recalibration and Deduplication of Aligned Reads.
- Utilize Freebayes for Variant Calling.
- Perform Variant Calling Using Freebayes.
- Explain DRAGEN: A Germline Variant Detector.
- Identify Germline Variants Using DRAGEN.
- Explain Mutect2: A Somatic Variant Caller.
- Identify Somatic Variants Using Mutect2.
- Explain HaplotypeCaller: A Germline Variant Caller.
- Identify Germline Variants Using HaplotypeCaller.
- Describe VCFtools: A Toolkit for Variant File Formats.
- Perform Variant Calling Using Bcftools.
- Perform Variant Recalibration of Raw Variants.
- Perform Variant Filtration.
- Filter Out the Low-Quality Variants Using SnpSift.
- Perform Variant Annotation.
- Annotate Variants Using SnpEff for Prediction of Variant Effects.
- Utilize Variant Effect Predictor (VEP) for Elucidation of Novel Variants.
Section 3: Additional Downstream Analysis
Description: Students will learn about the downstream functional enrichment analysis. They will learn how to perform gene ontology analysis using topGO and enrichR, as well as pathways analysis using KEGG, PANTHER, and Reactome.
Learning Outcomes: Upon completion of this section, students will be able to:
- Explain the Downstream Functional Enrichment Analysis.
- Perform Gene Ontology Analysis Using topGO & enrichR
- Perform Pathways Analysis Using KEGG, PANTHER, Reactome.
Section 4: Additional Lectures
Description: Students will have the access to additional lectures which will help them learn about various databases, and file formats so that they can easily retrieve the required biological data and perform whole-exome analysis.
Learning Outcomes: Upon completion of this section, students will be able to:
- Discuss ArrayExpress.
- Describe Gene Expression Omnibus Database.
- Explain NCBI Genomes & NCBI Assembly.
- Discuss Genome Reference Consortium.
- Retrieve an Entire Genome & Retrieval of SARS-CoV-2 Viral Genome.
- Perform Genome Assembly Retrieval and Analysis
- Explain BED.
- Describe GTF/GFF.
- Explain SAM/BAM.
Learning path
NGS & variant calling
- 1 Hands-on: NGS (Whole Exome Sequencing) Variant Calling Using Linux
- 2 Hands-on: NGS (Whole Exome Sequencing) Variant Calling Using Python
- 3 Cancer Genomics: NGS (Whole Genome Sequencing) Variant Calling Using Linux
- 4 Cancer Genomics: NGS (Whole Exome Sequencing) Variant Calling Using Linux · you're here
Not ready to enrol?
Get the free syllabus & course updates
We'll email you the full outline for this course plus a starter guide — no spam, unsubscribe anytime.
Tools & technologies you'll use
- Linux
- Bash / CLI
- BWA
- Bowtie
- Samtools
- BCFtools
- GATK
- IGV
Course Content
In-depth Introduction to NGS and Whole-Exome Sequencing for Cancer Genomics
-
In-depth Introduction to NGS and Variant Calling
20:12 -
Fundamentals of Whole Exome Sequencing: A Comprehensive Sequencing Approach
26:16 -
Cancer Genomics: Role of WES in Cancer Research
22:18 -
Haploid vs. Diploid Organisms for Variant Calling
07:26 -
Ploidy and Its Significance in Disease Research
14:58 -
A Bioinformatics Approach Towards Germline vs. Somatic Mutations
14:30 -
Single Nucleotide Polymorphisms: An Introduction to SNPs and Their Types
31:22 -
Structural Variations: Large Scale Variations in Genomes
08:55 -
Copy Number Variations: Role of CNVs in Diseases
10:29
Hands-on Whole-Exome Variant Calling
Additional Downstream Analysis
Additional Lectures
Earn a certificate
Add this certificate to your resume to demonstrate your skills & increase your chances of getting noticed.

Student Ratings & Reviews